ユーロフィンジェノミクス国内ラボで対応可能な、アプリケーションについてご紹介いたします。
▼ 基本的な解析 ▼ トランスクリプトーム解析 ▼ ゲノム解析
1.基本的な解析サービス
HiSeq 2500/2000 次世代シーケンシング: 2x125 bp ペアエンド解析
現在世界で最も使用されている次世代シーケンサーである、Illumina 社 HiSeq 2500/2000 での受託シーケンシングサービスを提供しています。当社の対応モジュールは、デファクトスタンダードになっている 2x125bp 解析です。これは、DNA断片である解析ライブラリの両端から 125bp ずつシーケンシングを行う、ペアエンドという解析方法です。配列データは、fastQ 形式でお届けします。
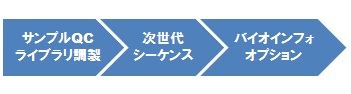
ライブラリ調製&シーケンスの一貫解析
DNA または total RNA をサンプルとしてお預かりして、解析に耐える品質か否かの確認(サンプルQC)を経て、ライブラリ調製→次世代シーケンシング解析の一貫した解析を、当社では推奨させていただいています。オプションとして、シーケンシング解析後のバイオインフォマティックス解析も承っております。
2. トランスクリプトーム解析
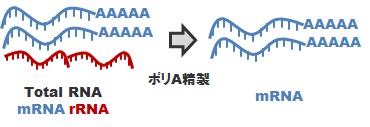
mRNA-Seqライブラリ調製: Poly-A精製
真核生物を対象とした標準ライブラリ調製方法です。total RNA をサンプルとしてお預かりして、polyA精製を行い、mRNAを精製します。この mRNA を cDNA 化し、所定のアダプタを付加することにより、mRNA-Seq (Strand Specific)ライブラリを調製します。
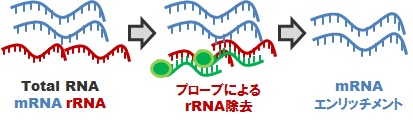
mRNA-Seqライブラリ: 細菌
rRNA 枯渇処理
原核生物(細菌)の場合、mRNA は原則として polyA 構造を持たないため、これを用いた mRNA 精製が困難です。かといって精製を行わないでシーケンシングをしても、rRNA配列ばかりを読んでしまうことになります。細菌トランスクリプトーム解析を実現するためには、予めプローブによる rRNA 枯渇処理を行ってから、cDNA 化、ライブラリ調製を行う必要があります。生物種により相性がありますので、予めご相談ください。なお、実際の rRNA 枯渇の程度については、当社で保証することはできません。
ヒト・マウス・ラットの total RNA サンプルからも、rRNAを効率よく除去することができます。断片化が進んだFFPEサンプルのrRNA除去に有効なだけではなく、ポリA精製によって失われていた、ポリA末端を持たないトランスクリプトをシーケンス解析対象とすることが可能となります。詳細については、お問い合わせください。
mRNA-Seq データのマッピング&遺伝子発現量解析
対象生物の遺伝子配列を参照配列にして、mRNA-Seq のシーケンシングリードデータをマッピング(貼り付け)し、マッピング結果を bam 形式ファイルでお届けします。遺伝子参照配列に張り付いたリードの数と、遺伝子の長さなどを勘案した RPKM (Reads Per Kilobase of exon per Million mapped sequence reads) 法などを用いれば、遺伝子発現量の正規化も可能です。RPKM 法などの計算を行いやすいように、データはエクセルなどで加工可能なcsv形式ファイルでお届けします。
mRNA-Seq データのマッピング&変異解析
mRNA-Seq データのマッピング結果からも、ゲノムリシーケンシング同様に、参照配列に対する変異解析(バリアント・コーリング)を行うことも可能です。ただし、cDNA 合成時にエラーが入りやすいために、ゲノム解析よりも精度が低く、データ解釈が難しくなる場合がありますので、ご注意ください。変異解析結果は、vcf 形式ファイルでお届けします。
3. ゲノム解析
ゲノム ショットガン ライブラリ調製: 300 bpインサート
ゲノムDNAをサンプルとしてお預かりて調製する、標準のゲノムショットガンライブラリとなります。物理的切断によりゲノムDNAを約 300bp の大きさに切断し、所定のアダプタを付加することにより、ゲノムショットガンライブラリを調製します。HiSeq 2500/2000 2x125bp 解析では、インサート長平均 300bp のゲノムインサートの両端 125bp をシーケンシングします。
ゲノム ショットガン ライブラリ調製: 500, 700 bpインサート
標準では 300bp のインサートサイズのゲノムショットガンライブラリですが、500bp, 700bp のインサートサイズのカスタムサイズのゲノムショットガンライブラリを調製することも可能です。これらのインサートサイズが異なるライブラリをシーケンシング解析することで、de novo アセンブルの精度向上の可能性が高まります。
ヒト エキソームライブラリ調製
ヒトゲノムは 3Gb と非常に大きなものですが、ヒトの全エキソン(エキソーム)は僅か 50Mb 程度です。ヒト エキソーム解析では、まず全エキソンに対するプローブを用いて全ゲノムからエキソン部分のみをキャプチャー濃縮し、その濃縮産物からライブラリを調製します。解析対象を全ゲノムから全エキソンに絞って解析することにより、コストを抑えることができるので、その分サンプル数を増やすような研究計画を立てることもできます。
ゲノムシーケンスデータのマッピング&変異解析
対象生物の既知ゲノム配列を参照配列にして、ゲノムリシーケンシングのリードデータをマッピング(貼り付け)し、マッピング結果を bam 形式ファイルでお届けします。このマッピング結果から、参照配列に対する変異解析(バリアント・コーリング)を行うことが可能です。各解析サンプルと参照配列との間の変異解析結果は、vcf 形式ファイルでお届けします。この vcf ファイルには、変異部位候補の染色体名、位置、ホモ/ヘテロ別などの簡易なアノテーション情報が記載されています.
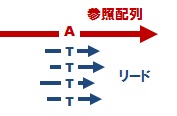
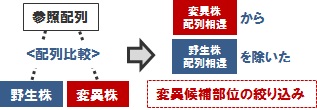
変異部位の絞り込み解析
上述の「標準のマッピング&変異解析」では、各解析サンプルと参照配列間の 「1対1」 の比較による変異部位候補のリストアップを行います。しかし実際には、解析を行った親株(野生株)と参照配列との間にも配列の相違があり、この相違の分はバックグラウンドとして、変異部位候補からまず除く必要があるでしょう。このような、ご要望に応じた変異部位の絞り込み解析もオプション解析として、ご要望に応じて実施します。結果は、vcf 形式ファイルでお届けします。
変異部位のアノテーション付与解析
上述の 「標準のマッピング&変異解析」 では、ミニマムなアノテーション情報(染色体名, 位置, ホモ/ヘテロ別など)のみが付与されます。オプションの 「変異部位のアノテーション付与サービス」 では、変異候補部位の詳細なアノテーション付与にも対応し、遺伝子名や SNP ID の付与だけでなく、変異候補部位が遺伝子領域であればエキソン内であるか…エキソン内であればアミノ酸変異を伴う変異であるか…といった詳細な情報出力に対応します。この情報を用いれば、数ある候補部位からアミノ酸変異を伴う変異のみに絞り込む、といったことも可能となります。結果は、vcf 形式ファイルでお届けします。